Tutorial : single vacancy in Silicon
This tutorial describes how to set-up a kART simulation of the evolution of a Stillinger-Weber crystalline silicon with a vacancy.
Problem
We are interested in studying the diffusion of a vacancy in crystalline Si at room temperature. We want to focus the attention on atoms surrounding the vacancy. We are not interested in possible events that could involve atoms in a perfectly crystalline environment as those events will be high in energy and of little relevance at room temperature.
System
We first need a starting configuration. In this tutorial, we pose that you already know how to generate such a configuration in the lammps format. For more information, see the lammps manual.
We start therefore with the file conf.sw, which describes a system with 511 atoms (and one vacancy) in a 21.72 A x21.72 A x 21.72 A crystalline Si box with periodic boundary conditions.
Box of Si with a vacancy
511 atoms
1 atom types
0.0 21.72 xlo xhi
0.0 21.72 ylo yhi
0.0 21.72 zlo zhi
0.0 0.0 0.0 xy xz yz
Atoms
1 1 0.00000000 0.00000000 0.00000000
2 1 2.71500000 2.71500000 0.00000000
3 1 0.00000000 2.71500000 2.71500000
...
510 1 17.64750000 20.36250000 20.36250000
511 1 20.36250000 17.64750000 20.36250000
kART uses the LAMMPS format only to ensure that the ordering of the atomic species is done properly. Atomic position in kART are read from its own file. This is why it is necessary to convert the file into the kART xyz format. This can be done in two steps:
Header
Consists of 3 times : the first one the id of the step, the second one the total energy and the third one, the box site.
Create a file name “511.conf” with the following first three lines
run_id: 0
total energy : 0.0000
21.72 21.72 21.72
Atomic positions
Atomic positions are given according to a simple 4 columm formats, including : atomic type, x, y and z positions.
From the lammps configuration file (
conf.sw), extract with :
% tail -n 511 conf.sw | awk '{print " " $2 " " $3 " " $4 " "$5}' >> 511.conf
Setting up the in.lammps file
Since we are using LAMMPS to compute forces and energy, it is necessary to set-up the file.
All lammps needs is (i) the definition the potential and (ii) a configuration with atoms ordered as in kART ; the position file does not need to be updated as the system evolve.
For the system of interest here, we define :
log log.lammps # This file can become large, comment out unles you need to debug
units metal # defines the units used, these have to be the same as for kART
atom_style atomic
atom_modify map array # This line is ESSENTIAL as it allows kART to update the atomic
# positions in lammps
read_data conf.sw # reads the box size and atomic position in the configuration file
# this file does not have to be updated as the system evolves
mass 1 28.0855 # atomic masses
# Definition of the potential
pair_style sw
pair_coeff * * Si.sw Si
neighbor 0.0 bin
neigh_modify delay 0 every 1 check no
It is possible to test lammps at this moment, simply by calling :
% lmp < in.lammps
If everything works, you should see something like :
LAMMPS (29 Sep 2021 - Update 2)
Reading data file ...
triclinic box = (0.0000000 0.0000000 0.0000000) to (21.720000 21.720000 21.720000) with tilt (0.0000000 0.0000000 0.0000000)
1 by 1 by 1 MPI processor grid
reading atoms ...
511 atoms
read_data CPU = 0.003 seconds
Total wall time: 0:00:00
Setting up the KMC.sh file
THe KMC.sh file can define a large number of parameters. Many of them have default values, but it is useful to go over the process.
Description of the system
The system must be described, including the system size, the configuration and others. These are self-explanatory.
setenv NUMBER_ATOMS 511 # The total number of atoms
setenv SIMULATION_BOX 21.72 # The size of the simulation box (x, y and z)
setenv NSPECIES 1 # The number of different atom types (default: 2)
setenv ATOMIC_SYMBOLS "Si" # The list of elements
setenv INI_FILE_NAME '511.conf' # The file name containing the intial configuration
setenv ENERGY_CALC LAM # choose between EDP or SWP or SWA or LAM (Lammps)
setenv INPUT_LAMMPS_FILE 'in.lammps' # LAMMPS input file when using LAM to calculate the forces
setenv UNITS_CONVERSION 'metal' # Converts the energy and distance units provided by the force code
# into a desired value for ART - affects only the parameters in ART and
# kART units available are real , electron and si. Be very
# careful when choosing other units, you must change parameters of energy
# and distance (if it's not in Angstrom) in input files for LAMMPS
Details of the simulation
The next parameters define the nature of the simulation, including the temperature, the number of KMC steps or the maximum simulation time, and whether the simulation continues a previous one or uses a new or old catalogue.
setenv TEMPERATURE 500.0 # The simulated temperature in kelvin
We can either set a maximum number of KMC steps or defined a maximum siimulation time. If the two are defined, the code stops when the first occurence is matched.
setenv NBRE_KMC_STEPS 50 # The max number of KMC steps to be executed
setenv TOTAL_TIME 1.0 # The maximum simulation time in seconds
Finally, the code can continue a simulation or start a new one:
setenv RESTART_FILE "this_conf" # The file name used to continue a simulation from where it was
# last stopped
setenv RESTART_IMPORT .false. # Start a NEW simulation but with the current KMC event catalogue
# (events.uft and topos.list)
setenv NEW_CATALOGUE .false. # IF true, will continue simulation but will rebuild event
# catalogue from scratch
Setting up the topological parameters
The topological parameters define the size of the graph needed to differentes local environments. If the graph is too small, many different geometries will share the same topology, meaning that the topological id is not a proper descriptor. If the graph is too large, then multiple topologies will correspond to the same geometry. This does not cause problem, however, it add to the computing cost. For a crystalline environment with local or extended defects, graphs containing 50 to 70 atoms is sufficient (47 for Si, 65, for Fe in the examples we are providing). This should be a bit bigger for glasses and disordered systems (70 - 90 atoms).
Defining the TOPO_RADIUS, that sets of graph size
TOPO_RADIUS must be set between peaks in the radial distribution function (RDF) to avoid multiplying topologies that are basically the same (if the cut-off is too close to a peak, any fluctuation will create a different topology)
The figure below shows the RDF for the 511-atom crystalline Si with one vacancy. We see that a good cut-off after the 5th neigbhor is something like 6.15 A. This is what we will use here.
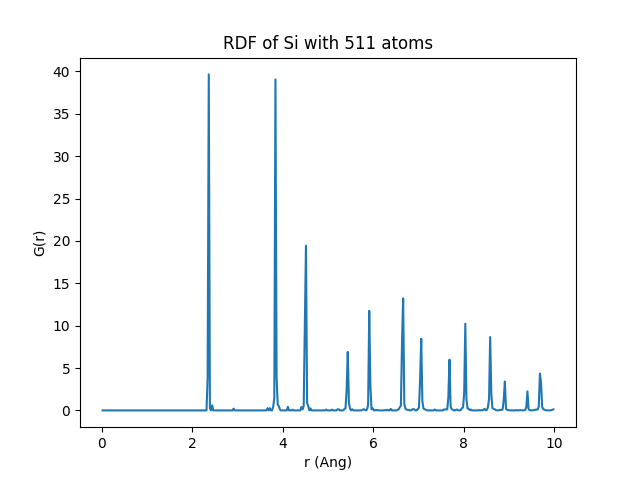
We also need to define first-neighbour cut-off to construct the graph. In Si, first neighbou distance is at 2.35 Å so a cut-off at 2.7 Å seems reasonnable. We allow for decreasing this cut-off when the same topology is found to correspond to more than one geometry. The minimum distance allowed, here, is 2.2 Å, a value that is never really used.
setenv TOPO_RADIUS 6.15 # radius for topology cluster
setenv MAX_TOPO_CUTOFF 2.7 # length-cutoff used by default to link two atoms
setenv MIN_TOPO_CUTOFF 2.2 # minimal length cutoff used when looking at
# secondary topologies
Defining the CRYST_TOPO_RADIUS and identifying the CRYST_TOPO_ID
To avoid generating event in crystalline environment, kART provides the option of ignore all atoms such environment. This is done, typically, with a shorter radius than for the TOPO_RADIUS as any atom with a crystalline environment in the first and second-neighbour shell is unlikely to be able to move easily.
Here, looking at the RDF above, we see that 4 Å is a good cut-off for CRYST_TOPO_RADIUS. We must now identify the TOPO_ID corresponding to this environment so that when it is found, the corresponding atom is ignored.
To identify this ID, we need to run kART with TOPO_RADIUS set at the shorter value and identify the topology given by this.
Define the TOPO_RADIUS
setenv TOPO_RADIUS 4 # radius for topology cluster corresponding to crystal cut-off
Launch kART
% srun -np 4 KMC.sh
Kill the job as soon as activation starts
... atom_label 511 crystalline: T 0.75356999999999996 : worker_0, sending task to worker with id: 1 ...
Identify the topo id. For this search for “listing topologies”
... ============================= Listing topologies in system: 973883( Multiplicity : 483, Crystallinity : F and master atom activity : T) this topology is ACTIVE 194271( Multiplicity : 24, Crystallinity : F and master atom activity : T) this topology is ACTIVE 736650( Multiplicity : 4, Crystallinity : F and master atom activity : T) this topology is ACTIVE known_tc, active_tc 3 3 ============================================
We see that the topo id 973883 is found 483 times, it is therefore associated with crystalline environment.
Set-up the CRYST_TOPO_RADIUS and ID in KMC.sh
setenv TOPO_RADIUS 6.15 # radius for topology cluster setenv MAX_TOPO_CUTOFF 2.7 # length-cutoff used by default to link two atoms setenv MIN_TOPO_CUTOFF 2.2 # minimal length cutoff used when looking at secondary topologies setenv CRYST_TOPOID 973883 # topo id of the crystalline-like topologies setenv CRYST_TOPO_RADIUS 4.0 # radius for crystal-like topologies (default: 4.0 A)
Basin parameters
kART manages basins, i.e. sets of states separated by low-energy barriers trapping the system, using teh bac-MRM approach as described in the documentation. The threshold barrier criterion for considered a two states in a basin, must be met by both direct and reverse barriers. If the barriers is higher in one direction, then the event is not included into the basin.
For a new system, it is better to first disable the basin. It is possible to ignore the low barriers, though,by keeping MIN_SIG_BARRIER:
setenv OSCILL_TREAT NONE # choose between BMRM, TABU or NONE
setenv MIN_SIG_BARRIER 0.1 # Max height of barrier and inv. barrier for an event to be considered inside
# a basin or, when BRMN is disabled, to ignore an event as insignificant.
The BRMN should be set only after characterization of the energy landscape.
Launching the simulation and looking at the results
The example used here generated 50 KMC steps in a simulation at 500 K.
% time srun -np 5 KMC.sh > kout
On a Mac Studio, this takes a minute or so:
real 1m2.385s
user 4m3.723s
sys 0m3.122s
Let us first consider Energy.dat, which presents the basic evolution of of the system.
# KMCs CPU time Old energy New energy barrier Total rate time step Sim time Selec Ev Init Topo Sad Topo Fin Topo
# **** *********** ********** *********** *********** ********** ********** *********** *********** *********** *********** ***********
0 0.0000E+00 0.0000 -2213.3483 0.0000 0.0000E+00 0.0000E+00 0.0000E+00 0 0 0 0
1 0.1041E+02 -2213.3483 -2213.3482 0.5100 0.1082E+06 0.2258E-05 0.2258E-05 1085412 728084 1202243 728084
...
49 0.6057E+02 -2213.3482 -2213.3482 0.5100 0.1082E+06 0.6842E-05 0.3440E-03 1085412 728084 1202243 728084
50 0.6109E+02 -2213.3482 -2213.3482 0.5100 0.1083E+06 0.1337E-04 0.3574E-03 1085412 728084 1202243 728084
We see that selected barrier is 0.51 eV (symmetric), corresponding to 2.2 µs time step, with the vacancy jumping from one site to the next, always with the same environment as shown by the initial and final topologies.
Similarly, Diffusion.dat shows the atomic diffusion for this run.
# Elapsed Time Sqr Displ. Sqr Displ. KMC step
# ************ ***Total*** Atom Si ********
0.00000000E+00 0.0000000 0.0000000 0
0.22584926E-05 2.9829400 2.9829400 1
0.47105136E-05 0.0000002 0.0000002 2
...
0.33719704E-03 96.4798959 96.4798959 48
0.34403920E-03 101.6722196 101.6722196 49
0.35740465E-03 96.4792121 96.4792121 50
Accepted configurations (min-sad-min-sad…) are shown in allconf for the full configuration and allconf_defect for the atoms surrouding the diffusion atom. These files are in a format compatible with Ovito.
The event catalogue, which contains information about the energy landscape at each step can be found in the EVLIST_DIR subdirectory. A separate file will all available events is given at for each KMC step. The file event_list_conf_1 shows all events found from the initial configuration. It contains 60 events presented as
#TypeId AtomId IniTopoId SadTopoId FinTopoId eventId Spec_id barrier inv_bar asy_ener true_rate inisad_dr inifin_dr refined
#****** ******* ******* ******* ******* ******* ******* ******* ********* ********* ********* ********* ********* *******
1 331 236619 1253516 1108930 421298 1 2.351305 0.085162 2.266143 3.161E-27 1.617366 1.973920 F
1 331 236619 723439 975910 1228917 4 2.956779 0.150814 2.805965 2.130E-37 2.481541 2.733755 F
...
1 489 728084 1202243 728084 1085412 1 0.509955 0.509897 0.000058 2.711E+04 0.864151 1.723820 T
1 489 728084 890611 236918 694158 3 2.883835 0.481198 2.402638 3.579E-36 2.050739 2.441887 F
...
1 495 236619 624389 296339 1223290 3 2.852846 0.494603 2.358243 1.187E-35 1.622654 2.169408 F
1 495 236619 1023068 296339 272701 2 2.854317 0.496067 2.358249 1.121E-35 1.635074 2.169133 F
The first column indicates the element type of the central atom, whose number is given in the next column. Topological id for initial, saddle and final configurations are then provided for information. The next two columns given the eventId, which is detailed generically in a file with the same number in EVENTS_DIR. Direct and inverse barriers are given, along with the energy difference between final and inital state, transition rate and the total displacement to the saddle and the final state.
The last column indicates whether the event has been reconstructed and its saddle point relaxed specifically (T) or not. Only events that have a probability equal or higher to the SPECIFIC_THRESHOLD (default 99.99 %) to be selected are reconstructed (specific events). Here, we see that only four events are reconstructed, all with a barrier of about 0.5 eV, corresponding to a first-neighbour vacancy jump. The configuration and details of these reconstructed specific events are presented in the SPEC_EVENTS_DIR directory. Where names indicate the central atom specific id and steps.